Frequently Asked Questions
🧬 EDS 101: What Is Ehlers-Danlos Syndrome?
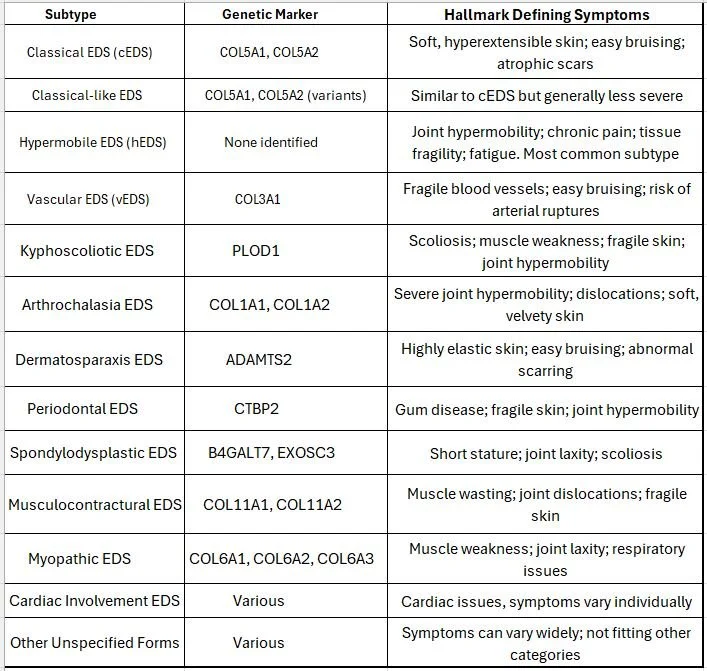
Ehlers-Danlos Syndromes (EDS) are a complex group of genetic disorders that affect connective tissues, especially collagen, which is essential for the strength and elasticity of skin, joints, blood vessels, and internal organs. When collagen is abnormal, it leads to many multisystem symptoms that are often overlooked or misdiagnosed. Currently, there are 13 recognized types of EDS, each with unique clinical features and genetic markers.
The table below summarizes these subtypes along with hallmark defining symptoms.
🧪 What Does It Mean That hEDS Is a Clinical Diagnosis?
Hypermobile Ehlers-Danlos Syndrome (hEDS) is particularly challenging to diagnose, as no specific genetic test currently exists. Instead, healthcare providers rely on a checklist of clinical symptoms and history.
Some diagnostic indicators include:
- Joint hypermobility, often quantified using the Beighton score.
- History of joint issues, such as frequent dislocations or instability.
- Signs of fragile tissue, such as soft skin, atrophic scarring, or hernias.
- Systemic symptoms affecting multiple body areas.
- Exclusion of other disorders with similar presentations.
Due to these complexities and the lack of a direct biomarker, hEDS is often misdiagnosed, particularly in settings where knowledge of connective tissue disorders is limited.
🧍♀️ What Are the Most Common Symptoms?
EDS affects multiple systems, often in overlapping ways. Common symptoms include:
- Joint hypermobility and frequent dislocations
- Chronic musculoskeletal and neuropathic pain
- Severe fatigue
- Stretchy, fragile skin; slow healing and easy bruising
- Gastrointestinal issues (IBS, reflux, etc.)
- Autonomic symptoms: dizziness, fainting, increased heartrate
- Headaches/Migraines and “brain fog”
- Pelvic floor dysfunction, endometriosis, or organ prolapse
🔄 What Conditions Frequently Overlap With EDS? (Comorbidities)
1. Postural Orthostatic Tachycardia Syndrome (POTS)
- Affects blood circulation. Standing up causes excessive heart rate increases, dizziness, or fainting. EDS-related vascular laxity may contribute.
2. Mast Cell Activation Syndrome (MCAS)
- Inappropriate mast cell release causes allergy-like symptoms (hives, swelling, GI issues). Seen frequently in EDS patients.
3. Gastrointestinal Disorders
- IBS and gastroparesis are common due to connective tissue dysfunction in the gut wall and nervous system regulation.
4. Temporomandibular Joint Dysfunction (TMJ)
- Connective tissue laxity makes the jaw joint more prone to instability, clicking, and pain.
5. Neurological Conditions
- Tethered cord, Chiari malformation, and Craniocervical Instability (CCI) can co-occur, potentially affecting spinal cord function and sensory processing.
6. Mental Health Conditions
- Depression and anxiety are common and may stem from chronic pain, systemic inflammation, and the difficulty of living with a misunderstood illness.
7. Sleep Disorders
- Non-restorative sleep, insomnia, and fatigue may result from autonomic issues, pain, or central sensitization.
❌ Common Misdiagnoses in EDS
Patients with EDS are often misdiagnosed with conditions that resemble their symptoms but do not address the underlying connective tissue disorder:
1. Fibromyalgia
- Overlapping chronic pain and fatigue often lead to this diagnosis without recognizing joint hypermobility.
2. Chronic Fatigue Syndrome (CFS)
- Shared features like debilitating fatigue and cognitive issues often cause confusion between CFS and EDS.
3. Anxiety Disorders
- Autonomic symptoms (like racing heart or dizziness) are misattributed to anxiety, obscuring the real cause.
4. Growing Pains (in children)
- Pediatric joint pain may be written off as “normal” growth, delaying early intervention.
5. Functional Neurological Disorders
- EDS-related neurological symptoms (e.g., tremors, weakness) can be misdiagnosed as psychogenic if tests appear normal.
6. Lupus (Systemic Lupus Erythematosus)
- Because both lupus and EDS can involve fatigue, joint pain, skin sensitivity, and multi-system effects. Some EDS patients are initially evaluated or treated for lupus, especially when lab tests are inconclusive or show nonspecific inflammation.
🚧 Challenges in the Field
Despite rising awareness, significant gaps in understanding and managing EDS remain:
1. Lack of a Biomarker for hEDS
- Diagnosis relies on clinical criteria due to the absence of a known gene, contributing to misdiagnosis and limited research inclusion.
2. Limited Medical Education
- Most healthcare providers receive minimal training on EDS, leading to delayed diagnoses and inappropriate referrals.
3. Research is Underfunded
- EDS may affect 1 in 500 people, yet research funding is limited. Few large studies exist, and treatment remains largely anecdotal.
4. Fragmented and Siloed Care
- EDS affects multiple systems, but care is often split among uncoordinated specialists. Patients are left to connect the dots themselves.
5. Medical Dismissal and Gaslighting
- Especially common among women, many patients are told their symptoms are psychological or exaggerated.
6. Misinterpretation as Child Abuse
- In children, symptoms like bruising and joint instability are sometimes mistaken for signs of abuse, leading to investigations and trauma.
7. The Double-Edged Sword of Social Media
- While platforms like TikTok raise awareness, they can also spread misinformation. Some doctors now dismiss patients who mention EDS, assuming they’re self-diagnosing based on internet trends.
💡 Why It Matters
Current estimates suggest hypermobile EDS might impact 1 in 500 individuals (668,000 people in the US alone), making it one of the more commonly undiagnosed and misdiagnosed conditions in healthcare.
Early diagnosis and intervention, especially in childhood, can prevent injuries, improve quality of life, and reduce long-term disability.
🧭 Conclusion
Understanding and recognizing EDS in its many forms is crucial for guiding affected individuals toward proper care. By fostering awareness, educating providers, and supporting more research, we can move toward a future where patients are seen, supported, and believed.
References
Bowen JM, et al. Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet. 175(1):27-39. DOI: 10.1002/ajmg.c.31548. → This paper discusses the clinical features, diagnostic criteria, and therapeutic strategies for managing classical EDS.
Malfait, F., et al. (2017). The 2017 International Classification of the Ehlers–Danlos Syndromes. Am J Med Genet C Semin Med Genet, 175(1):8-26. DOI: 10.1002/ajmg.c.31552. → This article provides an updated classification of EDS, facilitating recognition and management.
Karthikeyan, R., & Venkat-Raman, D. (2018). Hypermobile Ehlers–Danlos syndrome and pregnancy. Obstet Med, 11(4):160-165. DOI: 10.1177/1753495x18754577. → Cited for practical considerations regarding pregnancy-related health issues in hypermobile EDS.
Dhingra, K., et al. (2021). Gastrointestinal medication burden among persons with the Ehlers‐Danlos syndromes. Neurogastroenterology & Motility, 33(3):e13850. - DOI:10.1111/nmo.14077. → This study highlights the gastrointestinal complications commonly reported in individuals with EDS.
Vos A, Burns KM. (2021) Pediatric Innominate Artery Pseudoaneurysm Rupture in Vascular Ehlers-Danlos Syndrome: A Case Report. Clin Pract Cases Emerg Med. 5(2):226-229. - DOI: 10.5811/cpcem.2021.3.51787. → Discusses critical vascular complications in EDS from a case management perspective.
Chun, H. Y., et al. (2011). Type IV Ehlers-Danlos Syndrome: A Surgical Emergency? A Case of Massive Retroperitoneal Hemorrhage. Open Cardiovascular Medicine Journal, 5:21-25. DOI: 10.2174/1874192401105010210. → This paper documents surgical challenges associated with vascular EDS.
Semyachkina AN, et al. (2021) Ehlers-Danlos syndrome kyphoscoliotic type 2 caused by mutations in the FKBP14 gene: an analysis of five cases. F1000Res., 10:502. DOI: 10.12688/f1000research.52268.1. → Explains the complexities in the management of kyphoscoliotic EDS.
van Dijk FS, et al. (2024) Clinical diagnosis of the monogenic Ehlers-Danlos syndromes. Med Genet. 36(4):225-234. DOI: 10.1515/medgen-2024-2060. → Offers an extensive overview of clinical presentations and challenges across various types of EDS.
Kim, S. J., et al. (2020). Appendiceal torsion in Ehlers-Danlos syndrome: A case report of a rare phenomenon in a rare disease. International Journal of Surgery Case Reports, 75:133-136. DOI: 10.1016/j.ijscr.2020.06.084). → Illustrates unique surgical challenges related to EDS.
Scheper, M., et al. (2015). Chronic Pain in Hypermobility Syndrome and Ehlers–Danlos Syndrome (Hypermobility Type): It is a Challenge. Journal of Pain Research, 8:93-100. DOI: 10.2147/jpr.s64251. → Explores chronic pain issues within the context of hypermobility EDS, highlighting comorbidities.
Wright, K., et al. (2023). Delivery Outcomes and Postpartum Readmissions Associated with Ehlers–Danlos Syndrome. American Journal of Perinatology. [DOI: 10.1055/a-2185-4149](DOI: 10.1055/a-2185-4149). → Addresses the obstetric risks and challenges faced by women with EDS.
Cosare, A., et al. (2024). Multisystem Involvement in a Pediatric Patient With Hypermobile Ehlers-Danlos Syndrome: A Case Report of the Diagnostic Complexity and Management Challenges. Cureus, 16(4):e62083. DOI: 10.7759/cureus.62083. → Discusses complexities in diagnosis and management in pediatric patients with EDS.
Scicluna K, et al. (2022) Hypermobile Ehlers-Danlos syndrome: A review and a critical appraisal of published genetic research to date. Clin Genet. 101(1):20-31. DOI: 10.1111/cge.14026. → underscores the complexities of diagnosing hEDS, the need for more robust genetic research, and the importance of a standardized approach in clinical practice to ensure timely and accurate diagnosis for individuals affected by this condition.
Beighton P, Solomon, et al. (1973) Articular mobility in an African population. Ann Rheum Dis. 32(5):413-8. DOI: 10.1136/ard.32.5.413 → This paper introduced the now widely used 9-point Beighton scoring system for evaluating generalized joint hypermobility in population studies, which has since been adapted in clinical diagnostic criteria for hEDS and other connective tissue disorders
💬 Other FAQ
What is The Connective Issues?
Connective Issues is a platform built at the intersection of science, lived experience, and system-level frustration. Created by a rare disease scientist and patient, it brings clarity to the often confusing world of Ehlers-Danlos Syndromes (EDS), misdiagnosis, and invisible illness. Through research, storytelling, and advocacy, we’re working to unravel the medical disconnects that leave too many people without answers and offer tools that help patients and practitioners alike reconnect the dots.
What is the Mission of Connective Issues?
The mission is to bridge the gap between science, medicine, and real-life patient experience. We believe no one should have to suffer for years without answers simply because their symptoms don’t fit inside a medical checklist. Connective Issues exists to educate, advocate, and build something better, together.
What is the Vision?
A world where EDS and other invisible conditions are no longer dismissed, misdiagnosed, or misunderstood. We envision a diagnostic landscape shaped by interdisciplinary research, empathetic listening, and systems that catch patients before they fall through the cracks.
Who is behind this effort?
This platform is led by Jaime Prout, a PhD molecular geneticist, former biotech researcher, mom of four, and a patient with hEDS. After experiencing the all-too-common diagnostic odyssey firsthand and through family members, Jaime set out to create what she wished existed: a space where researchers, advocates, patients, clinicians, and providers could actually work together to elicit change.
What is the survey for?
The current EDS Community Survey is focused on collecting detailed insights from patients navigating Ehlers-Danlos Syndrome and related conditions. It explores diagnostic timelines, symptom patterns, misdiagnoses, and care experiences, all to identify recurring issues and unmet needs.
This data will directly support the upcoming book and help shape future outreach, education, and advocacy. Additional surveys for caregivers, clinicians, and other community members may follow as the project expands.
[Take the Survey → https://forms.gle/aEWyYTtKoJo5tpeo8]
What is The EDS Voices Project?
The EDS Voices Project is an ongoing 100-person interview series collecting deep, diverse insights from across the Ehlers-Danlos and rare disease landscape. Each conversation explores the lived experiences, frustrations, breakthroughs, and ideas of patients, caregivers, clinicians, researchers, and advocates.
The goal is to move beyond anecdotes and to document patterns, expose system-wide gaps, and highlight solutions that can improve recognition, diagnosis, and care. These interviews will directly shape the upcoming Connective Issues book and serve as a foundation for future awareness and education efforts.
If you’ve lived it, treated it, studied it, or fought for someone with it, contact me! We want your voice in the story.
What is this about a book?
The Connective Issues book will be a science-informed, patient-driven exploration of Ehlers-Danlos Syndrome (EDS) and the broader challenges of complex, often-overlooked conditions. Written by Jaime M. Prout, PhD, a molecular geneticist and hEDS patient, the book will combine personal narrative, clinical insight, and current research to examine why diagnostic delays are so common and what can be done to improve outcomes.
In addition to sharing lived experience, the book breaks down the underlying science and physiology of EDS to help both patients and clinicians better understand how to identify, diagnose, and manage the condition. It aims to serve as both a resource and a catalyst for change across healthcare and patient communities.
What kinds of research are you doing?
We’re blending narrative research with data-driven insights to uncover patterns in diagnostic delays, misdiagnosis, comorbidity trends, and systemic gaps. This includes:
Patient-reported experiences
Scientific literature synthesis
Interviews with clinicians, researchers, and advocates
Case study aggregation
If you’re a researcher, provider, or EDS expert, get in touch Let’s collaborate!
How can I help?
Glad you asked. Here are a few ways:
Take the survey and share it with others
Subscribe to the blog for new science breakdowns and stories
Share your story or get interviewed
Invite us to collaborate with your organization
Coming Soon!
Follow and engage on social media (@connectiveissues)
Buy from the store, a portion of proceeds support the book and outreach efforts
How can I get in touch?
You can reach out directly via the Contact Page, email, or social media. Whether you’re a patient, provider, researcher, or ally, your message matters. Collaboration is the most important part of the connective tissue holding this project together.
Is this a nonprofit or a business?
Connective Issues is currently a patient-led, independent project, not a formal business or nonprofit. As it grows, the goal is to evolve into a sustainable platform for research, education, and advocacy. Every survey, share, and store purchase (coming soon!) helps support that mission.